欧盟医疗器械制造的重大监管变更
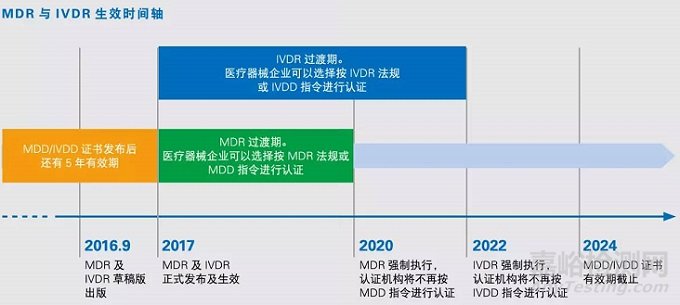
2018-04-18 13:34:53医疗器械制造商很快将面临一项监管架构的重大变更,该监管架构过去几十年来规范着欧盟的市场准入机制。欧盟委员会、欧洲理事会和欧洲议会正在就新的医疗器械规范(MDR)开展三方的最终磋商。一旦最终获得审批通过,MDR将替代欧盟现行医疗器械指令(93/42/EEC)和有源可植入医疗器械指令(90/385/EEC)。
针对医疗器械与有源可植入医疗器械,拟定的MDR与欧盟现行指令在多个方面有重要的不同。
拟定规范中最为显著的变更包括:
·产品范围扩大——MDR中针对医疗器械与有源可植入医疗器械的定义范围有了明显的扩大,包含了可能不具医疗预期用途的器械,如美瞳和整形植入器械与材料。另外,“预测”疾病或其他健康状况的器械也将属于规范的覆盖范围。
·更加严格的临床证据——MDR将要求医疗器械制造商开展临床性能研究,并提供该器械的与其风险程度相适应的安全与性能临床证据。医疗器械制造商还需收集并保存上市后的临床跟踪数据,作为持续评估潜在安全风险的部分依据。
·确定“具有资质的人员”——医疗器械制造商应当在组织内部确定至少一名人员,对组织满足MDR的所有要求负有最终责任。组织机构需记录该人员与各项工作相关的资质信息。
·执行器械唯一标识——拟定MDR要求使用医疗器械唯一标识(UDI)机制。该要求预计将增强制造商和有关当局通过供应链追溯医疗器械的能力,同时益于对存在安全风险的器械及时、高效地召回。除此以外,欧洲医疗器械数据库(Eudamed)预计会扩展,为获得已通过审批的医疗器械的相关信息提供更加高效的获取途径。
·严格的上市后监管——MDR将赋予公告机构更多的上市后监管的权利。飞行检查,产品抽查和产品测试将强化欧盟的执法机制,有助于降低不安全器械风险。很多情况下,还需要医疗器械制造商进行年度安全与性能汇报。
·规范——MDR计划允许欧盟委员会或专家小组公布通用规范,届时制造商和公告机构需要将其纳入到考虑范畴。这些通用规范将与协调标准及当前工艺发展水平并行存在。
